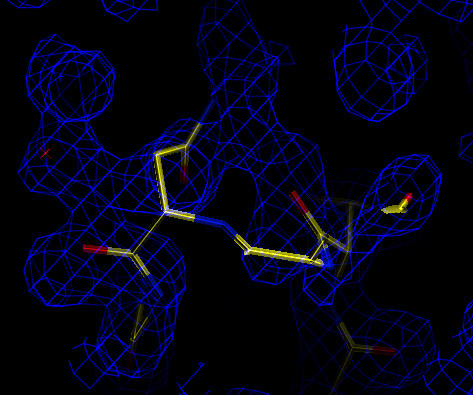
Incorrect conformation for loop at residue A200.
The model statistics are checked (as at the start of refinement) in order to check the model geometry and fit to the experimental diffraction data. In this example the CNS task file model_stats_final.inp is used to analyse the model geometry and diffraction statistics:
cns_solve < model_stats_final.inp > model_stats_final.out [2 minutes]
The output listing file (model_stats_final.list) contains a variety of information which is self-explanatory. Important things to note are:
R-values:
=================================== summary ==================================
resolution range: 500 - 1.8 A
R-values:
initial r= 0.3157 free_r= 0.3124
after B-factor and/or bulk solvent correction r= 0.2568 free_r= 0.2812
R-values versus resolution:
================================= R-values ===================================
=======> R-values with |Fobs|/sigma cutoff= 0.0
Test set (test = 1):
#bin | resolution range | #refl |
1 3.88 500.01 354 0.2481
2 3.08 3.88 354 0.2798
3 2.69 3.08 375 0.3010
4 2.44 2.69 394 0.3350
5 2.27 2.44 349 0.2734
6 2.13 2.27 384 0.2835
7 2.03 2.13 349 0.2864
8 1.94 2.03 348 0.2784
9 1.86 1.94 374 0.2577
10 1.80 1.86 393 0.2865
Working set:
#bin | resolution range | #refl |
1 3.88 500.01 3463 0.2479
2 3.08 3.88 3468 0.2605
3 2.69 3.08 3422 0.2829
4 2.44 2.69 3431 0.2775
5 2.27 2.44 3444 0.2655
6 2.13 2.27 3416 0.2375
7 2.03 2.13 3438 0.2412
8 1.94 2.03 3443 0.2381
9 1.86 1.94 3400 0.2468
10 1.80 1.86 3323 0.2535
This distribution of R-values is reasonable - there is no dramatic increase in R-value (in particular free R-value) as resolution increases. If there were resolution shells with R-values significantly higher than the rest this might indicate possible problems with the data processing (ice rings for example). However, the R-values indicate the fit to the experimental data and should not be manipulated by removing data - in particular by the use of sigma cutoffs to exclude weak data.
The overall geometry:
rmsd bonds= 0.005513 with 0 bond violations > 0.05 rmsd angles= 1.20416 with 3 angle violations > 8.0 rmsd dihedrals= 23.30213 with 0 angle violations > 60.0 rmsd improper= 0.81570 with 7 angle violations > 3.0
These overall statistics look very good. It is worth checking the outlying angle and improper deviations in the detailed geometry analysis (below).
The geometry in detail:
================================= geometry =================================== =======> bond violations (atom-i |atom-j ) dist. equil. delta energy const. =======> angle violations (atom-i |atom-j |atom-k ) angle equil. delta energy const. (A 198 N |A 198 CA |A 198 C ) 99.687 111.200 -11.513 10.009 247.886 (B 182 N |B 182 CA |B 182 C ) 101.078 111.200 -10.122 7.737 247.886 (B 198 N |B 198 CA |B 198 C ) 102.638 111.200 -8.562 5.536 247.886 =======> improper angle violations (atom-i |atom-j |atom-k |atom-L ) angle equil. delta energy const. period (A 120 N |A 120 CA |A 120 CD |A 119 C ) -3.715 0.000 3.715 0.420 100.000 0 (A 138 N |A 138 CA |A 138 CD |A 137 C ) -7.291 0.000 7.291 1.620 100.000 0 (A 168 CG |A 168 CD1 |A 168 CD2 |A 168 CB ) 3.727 0.000 -3.727 3.174 750.000 0 (A 206 CA |A 206 N |A 206 C |A 206 CB ) 32.042 35.264 3.222 2.372 750.000 0 (B 120 N |B 120 CA |B 120 CD |B 119 C ) -4.042 0.000 4.042 0.498 100.000 0 (B 138 N |B 138 CA |B 138 CD |B 137 C ) -5.320 0.000 5.320 0.862 100.000 0 (B 161 CA |B 161 N |B 161 C |B 161 CB ) 32.220 35.264 3.044 2.117 750.000 0 =======> dihedral angle violations (atom-i |atom-j |atom-k |atom-L ) angle equil. delta energy const. period
Specific problems with the model geometry can be identified here. The angle deviation all occur at N-CA-C angles. Based on high resolution structures this angle is flexible, so small deviations for some of these angles are tolerated.
After analysis of the geometry and fit to the experimental data electron density maps should be checked. Cross-validated, sigma-A weighted 2Fo-Fc and difference maps are calculated:
cns_solve < model_map_final_1fofc.inp > model_map_final_1fofc.out [30 seconds]
cns_solve < model_map_final_2fofc.inp > model_map_final_2fofc.out [1 minute]
If you have mapman installed, you can use the command
map_to_omap *.map
to convert the CNS maps to a format which can be read into
O. In O, enter @omac to read in the current
model and map.
Analysis of the difference map shows that some ordered water molecules are still missing (not shown). These can be placed either manually or by further rounds of automated water picking. Analysis of the 2Fo-Fc map indicates a loop in both molecules that is incorrectly placed. This can be manually rebuilt and further refinement carried out:
|
| Cross-validated Sigma-A weighted 2Fo-Fc map (at 1.5 sigma). Incorrect conformation for loop at residue A200. |