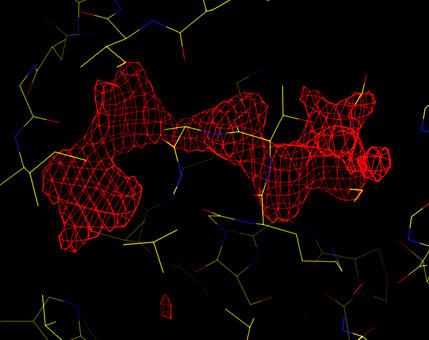
The density for the missing ATP molecule is clearly seen.
A difference map is calculated in order to locate either missing parts of the model or incorrectly placed atoms. The major error in the model at this point is the absence of an ATP molecule which we think is bound to the molecule. We hope that this will be apparent in the difference map when contoured at positive sigma. Here a cross-validated, sigma-A weighted, phase combined difference map is calculated using the CNS task file model_map_1fofc.inp:
cns_solve < model_map_1fofc.inp > model_map_1fofc.out [50 seconds]
If you have mapman installed, you can use the command
map_to_omap *.map
to convert the CNS maps to a format which can be read into
O. In O, enter @omac to read in the current
model and map.
The map shows clear density for an ATP molecule and and associated magnesium ion. The positive density peak list generated (model_map_1fofc_positive.peaks) locates the 3 phosphate and 1 magnesium atoms (the top 4 peaks respectively). This peak list (which is in PDB format) is used to extract the coordinates for the magnesium atom. The electron density is used as a guide to manually fit an ATP molecule.
The 4th peak in the positive peak list:
ATOM 1848 PEAK PEAK 4 10.685 28.812 36.469 1.00 11.87 PEAK
The electron density map:
|
| Sigma-A weighted, phase combined difference map (at 5 sigma). The density for the missing ATP molecule is clearly seen. |
The fitted ATP molecule:
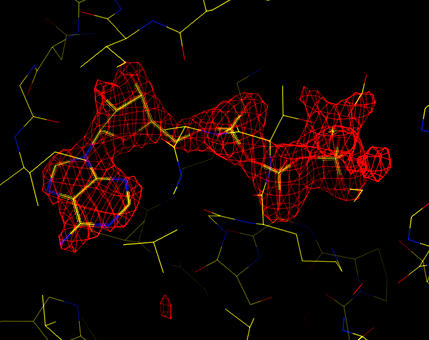 |
| Sigma-A weighted, phase combined difference map (at 5 sigma). The ATP molecule has been fitted manually into the density. |
In order to generate a new model with the ATP included we make a separate PDB file with the ATP and magnesium coordinates. The ATP coordinates can be obtained from a variety of sources, we used Gerard Kleywegt's HIC-Up database (http://welcome.to/hicup). The atom name and residue name for the magnesium ion must be changed manually. The ATP coordinate file looks like this:
ATOM 1 PG ATP 1 12.666 27.828 34.118 1.00 15.00 ATOM 2 O1G ATP 1 11.500 27.624 34.971 1.00 15.00 ATOM 3 O2G ATP 1 13.874 27.123 34.548 1.00 15.00 ATOM 4 O3G ATP 1 12.306 27.574 32.681 1.00 15.00 ATOM 5 PB ATP 1 12.257 30.747 34.155 1.00 15.00 ATOM 6 O1B ATP 1 11.240 30.777 35.237 1.00 15.00 ATOM 7 O2B ATP 1 11.791 30.936 32.766 1.00 15.00 ATOM 8 O3B ATP 1 13.108 29.368 34.276 1.00 15.00 ATOM 9 PA ATP 1 13.728 32.416 36.010 1.00 15.00 ATOM 10 O1A ATP 1 12.731 33.444 36.312 1.00 15.00 ATOM 11 O2A ATP 1 14.009 31.366 37.013 1.00 15.00 ATOM 12 O3A ATP 1 13.315 31.778 34.611 1.00 15.00 ATOM 13 O5* ATP 1 15.072 33.088 35.601 1.00 15.00 ATOM 14 C5* ATP 1 15.179 34.453 35.185 1.00 15.00 ATOM 15 C4* ATP 1 16.449 35.036 35.742 1.00 15.00 ATOM 16 O4* ATP 1 16.780 36.171 34.905 1.00 15.00 ATOM 17 C3* ATP 1 16.347 35.566 37.156 1.00 15.00 ATOM 18 O3* ATP 1 17.684 35.539 37.687 1.00 15.00 ATOM 19 C2* ATP 1 15.874 36.974 36.892 1.00 15.00 ATOM 20 O2* ATP 1 16.185 37.869 37.946 1.00 15.00 ATOM 21 C1* ATP 1 16.714 37.391 35.661 1.00 15.00 ATOM 22 N9 ATP 1 16.087 38.446 34.822 1.00 15.00 ATOM 23 C8 ATP 1 16.641 39.631 34.388 1.00 15.00 ATOM 24 N7 ATP 1 15.859 40.367 33.648 1.00 15.00 ATOM 25 C5 ATP 1 14.699 39.615 33.586 1.00 15.00 ATOM 26 C6 ATP 1 13.444 39.826 32.945 1.00 15.00 ATOM 27 N6 ATP 1 13.215 40.918 32.234 1.00 15.00 ATOM 28 N1 ATP 1 12.470 38.880 33.075 1.00 15.00 ATOM 29 C2 ATP 1 12.709 37.783 33.794 1.00 15.00 ATOM 30 N3 ATP 1 13.841 37.455 34.448 1.00 15.00 ATOM 31 C4 ATP 1 14.816 38.437 34.299 1.00 15.00 ATOM 32 MG+2 MG2 1 10.685 28.812 36.469 1.00 15.00
Note that the atom name for the magnesium ion has been changed to MG+2 and the residue name to MG2, these are the appropriate names for a magnesium 2+ ion. This information can be obtained from the CNS topology file CNS_TOPPAR:ion.top. It is not necessary to include a segid or chainid because in the subsequent generate stage we will rename the segid. This coordinate file is then used in combination with the protein coordinates to generate a new coordinate and molecular topology file for CNS. Here the CNS task file generate.inp is used:
cns_solve < generate.inp > generate.out [8 seconds]
In order to generate the molecular topology information and carry out refinement both topology and parameter information are required for the ATP molecule. These files can also be obtained from Gerard Kleywegt's HIC-Up database (http://welcome.to/hicup). For use in refinement the parameter file needed two modifications to allow different dihedral values from those present in the ATP molecule used to generate the topology and parameters. The force constants for the dihedrals around the PA O5* and C5 C4 bonds were set to 0 (to allow any dihedral value).
{ edit if necessary }
DIHEdral OX6 PX5 OX8 PX1 750.0 0 90.00 ! Nobs = 1 ... Value = 97.05
DIHEdral OX10 PX9 OX12 PX5 750.0 0 -60.00 ! Nobs = 1 ... Value = -64.65
DIHEdral OX11 PX9 OX12 PX5 750.0 0 60.00 ! Nobs = 1 ... Value = 68.21
DIHEdral OX13 PX9 OX12 PX5 750.0 0 180.00 ! Nobs = 1 ... Value = -176.43
DIHEdral OX10 PX9 OX13 CX14 0.0 0 180.00 ! Nobs = 1 ... Value = 179.62 <----
DIHEdral OX12 PX9 OX13 CX14 750.0 0 -60.00 ! Nobs = 1 ... Value = -68.35
DIHEdral OX13 CX14 CX15 OX16 750.0 0 60.00 ! Nobs = 1 ... Value = 63.80
DIHEdral OX13 CX14 CX15 CX17 0.0 0 180.00 ! Nobs = 1 ... Value = -178.18 <----
DIHEdral OX20 CX19 CX21 NX22 750.0 0 -90.00 ! Nobs = 1 ... Value = -83.44
The new coordinates are then refined further. It is important to first obtain reasonable estimates for the ATP and magnesium ion B-factors. This is done with group B-factor refinement. The ATP molecule and magnesium ion are each treated as a group. They are selected for refinement in the task file using:
{* select atoms in group 6 *}
{===>} group_6=(resname ATP or resname MG2);
The CNS task file bgroup_atp.inp
cns_solve < bgroup_atp.inp > bgroup_atp.out [4 minutes]
The B-factor refinement is followed by energy minimization of the
coordinates. Only 50 steps are performed as we will follow this with a
manual rebuild (see the next section). The CNS task file
minimize.inp
cns_solve < minimize.inp > minimize.out [7 minutes]
Script to run this tutorial