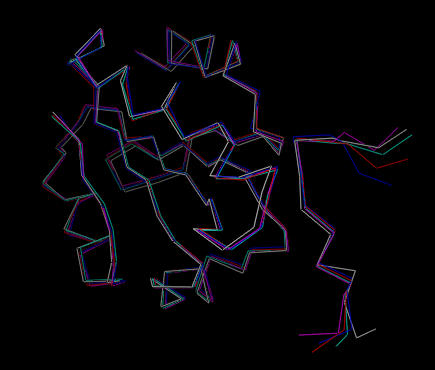
Here a structure is calculated using experimentally measured interproton distance estimates, hydrogen bonds and coupling-constant-derived dihedral angle restraints. This protocol uses ab initio simulated annealing starting from an extended template structure (see previous sections). The structure calculation is performed with the CNS task file anneal.inp.
cns_solve < anneal.inp > anneal.out [45 minutes]
In this case five trial structures were generated. An alternative approach would have been to only generated accepted structures, in which case the script would have run until five accepted structures had been generated. The five structures are quite similar:
|
| Five structures calculated from simulated annealing. |
A summary of the structure calculation is written at the top of each output PDB file:
REMARK The macromolecule has 118 residues REMARK Trial structure 1 of 5 structures REMARK Molecular dynamics scheme : torsion; torsion; cartesian; minimize REMARK High temperature dynamics : REMARK temp: 50000 steps: 1000 time(ps): 15 REMARK 1st cooling stage : REMARK temp: 50000->0 steps: 1000 time(ps): 15 temp step: 250 REMARK 2nd cooling stage : REMARK temp: 2000->0 steps: 3000 time(ps): 15 temp step: 25 REMARK a total 2000 steps of minimization REMARK VDW scale factors 0.1; 0.1->1; 1->4; 1 REMARK 2617 NOEs in 2 class(es) with scale factors of 150; 150; 150; 75 REMARK averaging function(s): sum, hbnd: sum REMARK 0 3-bond j-couplings in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 1-bond j-couplings in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 carbon chemical shifts in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 proton chemical shifts in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 diffusion anisotropy restraints in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 susceptability anisotropy restraints in 0 class(es) with REMARK scale factor(s) of NA REMARK 325 dihedral restraints with scale factors of 100; 200; 200; 400 REMARK 0 planarity restraints with a scale factor of NA REMARK NCS restraints not used. REMARK =============================================================== REMARK bond, angles, improp, vdw(<1.6), dihed REMARK violations : 2 8 5 4 49 REMARK RMSD : 0.0041 0.634 0.577 25.016 REMARK =============================================================== REMARK noe, cdih, coup, oneb, carb-a, carb-b, REMARK violations : 0 1 0 0 0 ----- REMARK RMSD : 0.022 0.729 0.000 0.000 0.000 0.000 REMARK 0.2/2 viol.: 7 6 0 REMARK =============================================================== REMARK dani, sani REMARK violations : 0 0 REMARK RMSD : 0.000 0.000 REMARK .2/.1 viol.: 0 0 REMARK =============================================================== REMARK Protons violations, rmsd REMARK all : 0 0.000 REMARK class 1: 0 0.000 REMARK class 2: 0 0.000 REMARK class 3: 0 0.000 REMARK class 4: 0 0.000 REMARK =============================================================== REMARK overall = 529.078 REMARK bon = 31.2461 REMARK ang = 204.65 REMARK imp = 47.5888 REMARK vdw = 133.24 REMARK harm = 0 REMARK noe = 91.3054 REMARK coup = 0 REMARK oneb = 0 REMARK carb = 0 REMARK prot = 0 REMARK dani = 0 REMARK sani = 0 REMARK cdih = 21.0478 REMARK ncs = 0 REMARK =============================================================== REMARK DATE: 8-Jul-99 18:18:29 created by user: paul REMARK VERSION:0.5 ATOM 1 CA MET 1 -9.804 10.394 -1.108 1.00 0.00 A ATOM 2 HA MET 1 -9.550 11.372 -0.727 1.00 0.00 A
The information about violations can be used manually to select acceptable structures. Instead the CNS task file accept.inp can be used to identify acceptable structures and to calculate an average conformation:
cns_solve < accept.inp > accept.out [2.5 minutes]
Information about accepted structures, violations and variations between structures is printed at the top of the average structure:
REMARK The macromolecule has 118 residues REMARK Accepted structures 5 of 5 structures REMARK 2617 NOEs in 2 class(es) REMARK averaging function(s): sum, hbnd: sum REMARK 0 3-bond j-couplings in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 1-bond j-couplings in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 carbon chemical shifts in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 proton chemical shifts in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 diffusion anisotropy restraints in 0 class(es) with REMARK scale factor(s) of NA REMARK 0 susceptability anisotropy restraints in 0 class(es) with REMARK scale factor(s) of NA REMARK 325 dihedral restraints REMARK 0 planarity restraints with a scale factor of NA REMARK NCS restraints not used. REMARK =============================================================== REMARK viol +/- sd rmsd +/- sd REMARK Bond : 2.00 +/- 0.000 0.0039 +/- 0.00017 REMARK Angl : 8.20 +/- 0.447 0.6118 +/- 0.0136 REMARK Impr : 2.60 +/- 1.342 0.4901 +/- 0.0494 REMARK VDW : 1.80 +/- 1.304 REMARK Dihed : 47.00 +/- 2.915 24.653 +/- 0.4696 REMARK =============================================================== REMARK viol +/- sd rmsd +/- sd REMARK NOE : 0.00 +/- 0.000 0.0200 +/- 0.0009 REMARK cdih : 1.00 +/- 0.000 0.6005 +/- 0.0711 REMARK J-coup : 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK Oneb : 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK =============================================================== REMARK viol +/- sd rmsd A +/- sd rmsd B +/- sd REMARK Carb : 0.00 +/- 0.000 0.000 +/- 0.000 0.000 +/- 0.000 REMARK =============================================================== REMARK Protons viol +/- sd rmsd +/- sd REMARK all : 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK class 1: 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK class 2: 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK class 3: 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK class 4: 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK =============================================================== REMARK viol +/- sd rmsd +/- sd REMARK dani : 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK sani : 0.00 +/- 0.000 0.0000 +/- 0.0000 REMARK =============================================================== REMARK overall = 441.00 +/- 44.01 REMARK bond = 27.36 +/- 2.41 REMARK angle = 190.47 +/- 8.58 REMARK improp = 34.63 +/- 7.37 REMARK vdw = 102.84 +/- 17.14 REMARK harm = 0.00 +/- 0.00 REMARK noe = 78.47 +/- 7.58 REMARK coup = 0.00 +/- 0.00 REMARK oneb = 0.00 +/- 0.00 REMARK carb = 0.00 +/- 0.00 REMARK prot = 0.00 +/- 0.00 REMARK dani = 0.00 +/- 0.00 REMARK sani = 0.00 +/- 0.00 REMARK cdih = 7.22 +/- 1.82 REMARK ncs = 0.00 +/- 0.00 REMARK =============================================================== REMARK rms around the mean for selection = 0.603 +/- 0.4288 REMARK rms around the mean for non-h atoms = 0.850 +/- 0.5029 REMARK =============================================================== REMARK pairwise rms REMARK atom selection: 0.920 +/- 0.2895 min: 0.550 max: 1.305 REMARK non-hydrogen : 1.203 +/- 0.1997 min: 0.924 max: 1.483 REMARK =============================================================== REMARK DATE:13-Jul-99 12:33:27 created by user: paul REMARK VERSION:0.5 ATOM 1 CA MET 1 -9.639 10.257 -0.958 1.00 0.70 A ATOM 2 HA MET 1 -9.301 11.261 -0.749 1.00 0.73 A