In this tutorial, MIR phases (see previous tutorial) are improved by solvent flipping and density truncation. In a preliminary step, the solvent content is estimated by using matthews_coef.inp.
cns_solve < matthews_coef.inp > matthews_coef.out [< 1 second]
The estimated solvent content shown in matthews_coef.list is between 0.32 and 0.35. density_modify.inp is modified accordingly by setting the solvent content parameter to 0.34.
cns_solve < density_modify.inp > density_modify.out [15 minutes]
The resulting output files are:
density_modify.list - listing file with statistics for
each modification cycle
density_modify.hkl - reflection file with new Hendrickson-Lattman
coefficients, and estimated applitudes for missing reflections.
density_modify.map - map file
density_modify.mask - mask file
Electron density maps before and after solvent flipping are computed
with modified copies of the fourier_map.inp task file. Note
that when making the map using the density modified phases the
reflection array produced by the density modification is used rather
than the native amplitudes. This reflection array contains estimated
amplitudes (and phases) for any missing reflections, these
reconstructed reflections can help improve the map, especially for
missing low resolution data.
cns_solve < fourier_map_mir.inp > fourier_map_mir.out [5 seconds]
cns_solve < fourier_map_dm.inp > fourier_map_dm.out [5 seconds]
If you have mapman installed, you can use the command
map_to_omap *.map
to convert the CNS maps to a format which can be read into O.
The refined heavy-atom positions in mir_phase_final.sdb (see previous tutorial) are converted to a PDB file for use in O.
cns_solve < sdb_to_pdb.inp > sdb_to_pdb.out [< 1 second]
In O, enter @omac to read in the maps and the heavy-atom
coordinates.
|
|
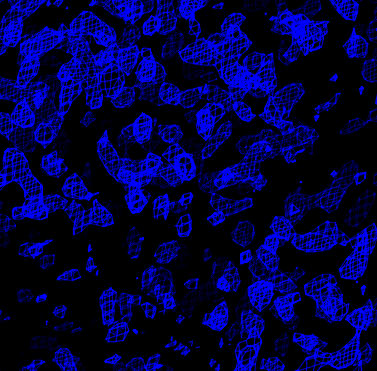
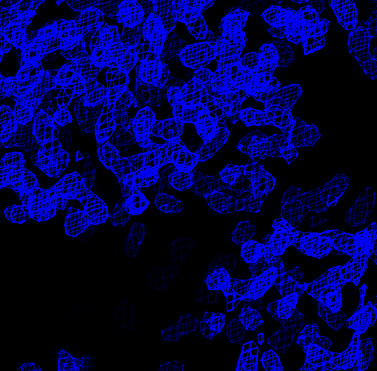
| Experimental MIR phased map (contoured at 1.5 sigma) | After solvent flipping (contoured at 1.5 sigma) |
At this point an interpretable electron density map is obtained. This allows the hand ambiguity of the heavy atom sites to be resolved. In the absence of anomalous diffraction data this ambiguity can only be resolved by visual analysis of the resulting electron density map. The handedness of secondary structure elements such as alpha-helices or the chirality of amino acids (given sufficient resolution of the data) will indicate if the heavy atom sites need to be inverted. If the sites do need to be be inverted the final MIR refinement job should be run after inverting the sites with the flip_sites.inp task file. The heavy atom coordinates for all derivatives will be inverted. The density modification should then be repeated.
Script to run this tutorial