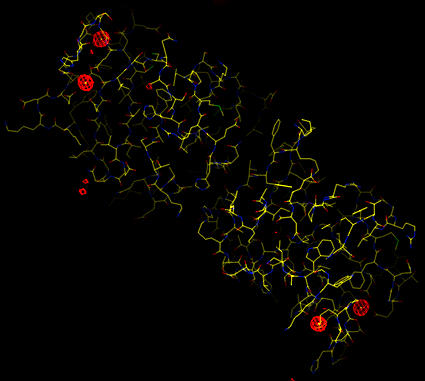
Four large positive peaks are clearly seen.
A difference map is calculated in order to locate either missing parts of the model or incorrectly placed atoms. The major error in the model at this point is the absence of several ytterbium ions which we know are bound in the calcium binding sites of the two molecules. We hope that these will be apparent in the difference map when contoured at positive sigma. Here a cross-validated, sigma-A weighted difference map is calculated using the CNS task file model_map_1fofc.inp:
cns_solve < model_map_1fofc.inp > model_map_1fofc.out [30 seconds]
If you have mapman installed, you can use the command
map_to_omap *.map
to convert the CNS maps to a format which can be read into
O. In O, enter @omac to read in the current
model and map.
The map shows 4 outstanding peaks (all above 20 sigma). These are clearly separated from the other difference density peaks in the map. This can be seen both by visual inspection of the map but also in the positive density peak list generated (model_map_1fofc_positive.peaks). This peak list (which is in PDB format) is used to add the ytterbiums to the model.
The positive peak list:
ATOM 1749 PEAK PEAK 1 17.491 36.603 50.078 1.00 33.53 PEAK ATOM 1750 PEAK PEAK 2 22.797 35.840 43.602 1.00 27.01 PEAK ATOM 1751 PEAK PEAK 3 1.547 -5.134 15.074 1.00 25.92 PEAK ATOM 1752 PEAK PEAK 4 8.461 -1.406 12.071 1.00 22.13 PEAK ATOM 1753 PEAK PEAK 5 30.817 23.796 35.520 1.00 6.50 PEAK ATOM 1754 PEAK PEAK 6 14.353 28.600 44.406 1.00 6.42 PEAK
The electron density map:
|
| Sigma-A weighted difference map (at 7 sigma). Four large positive peaks are clearly seen. |
In order to generate a new model with the ytterbium ions included we can merge the coordinates for the current model and the outstanding peaks from the peaks list. The atoms name, residue name and chain id for the ytterbium ions must be changed manually. The end of the new coordinate file look like this:
ATOM 1745 CG PRO B 220 -4.556 16.898 32.163 1.00 48.84 B ATOM 1746 C PRO B 220 -3.260 19.366 30.884 1.00 58.22 B ATOM 1747 O PRO B 220 -3.209 19.542 32.133 1.00 58.22 B ATOM 1748 OXT PRO B 220 -3.115 20.288 30.036 1.00 48.84 B ATOM 1749 YB+3 YB3 Y 1 17.491 36.603 50.078 1.00 33.53 ATOM 1750 YB+3 YB3 Y 2 22.797 35.840 43.602 1.00 27.01 ATOM 1751 YB+3 YB3 Y 3 1.547 -5.134 15.074 1.00 25.92 ATOM 1752 YB+3 YB3 Y 4 8.461 -1.406 12.071 1.00 22.13 END
Note that the atom name has been changed to YB+3 and the residue name to YB3, these are the appropriate names for a ytterbium 3+ ion. This information can be obtained from the CNS topology file CNS_TOPPAR:ion.top. The chain identifier has been changed to Y. It is not necessary to include a segid because in the subsequent generate stage we will any look at the chainid. This coordinate file is then used to generate a new coordinate and molecular topology file for CNS. Here the CNS task file generate_easy_yb.inp is used:
cns_solve < generate_easy_yb.inp > generate_easy_yb.out [2 seconds]
These new coordinates are then refined further. It is important to first obtain reasonable estimates for the ytterbium ion B-factors. This is done with group B-factor refinement. The ytterbium ions are each treated as a group (they will therefore undergo individual atomic B-factor refinement while the remainder of the model undergoes group B-factor refinement. The ytterbium ions are selected for refinement in the task file using:
{* select atoms in group 6 *}
{===>} group_6=(resname YB3);
The CNS task file bgroup_yb.inp. is used for the
refinement:
cns_solve < bgroup_yb.inp > bgroup_yb.out [2 minutes]
The B-factor refinement is followed by energy minimization of the coordinates. Only 50 steps are performed as we will follow this with a manual rebuild (see the next section). The CNS task file minimize.inp. is used:
cns_solve < minimize.inp > minimize.out [3.5 minutes]