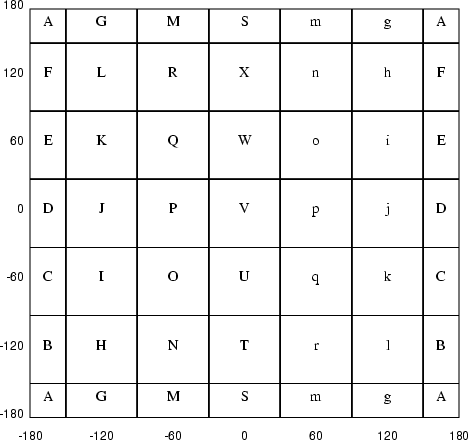
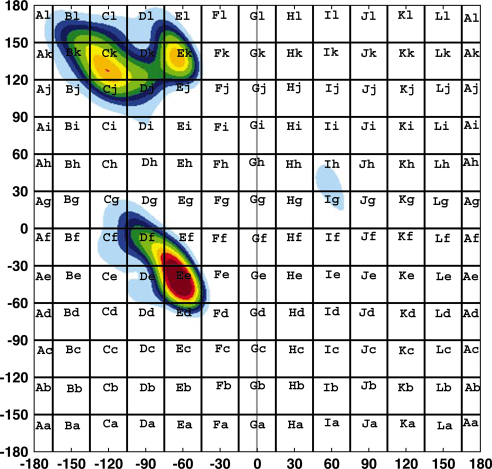
For example a residue with φ = -64° and ψ = -43° will be assigned a letter code of 'O' in he course grained grid and a letter code of 'Ee' in the fine grained grid.
Several methods have been developed for assigning a protein's secondary structure from information in a PDB file (i.e. x,y,z-coordinates). All methods use some combination of hydrogen-bonding patterns, backbone dihedral angles and idealized templates of α-helices, β-turns and β-strands. PROSS is one such method. In PROSS, secondary structure assignments are based solely on the backbone dihedral angles.
You may download a Python program to calculate secondary structure or use the web server.
For example a residue with φ = -64° and ψ = -43° will be assigned a letter code of 'O' in he course grained grid and a letter code of 'Ee' in the fine grained grid.